Did you know that roughly 90% of all prescriptions filled in the United States today are generic? These medications look different from their brand-name cousins-often cheaper, differently colored, or shaped-but they work exactly the same way inside your body. It is a system that has saved the healthcare industry over $1.7 trillion in the last decade alone. But how do manufacturers create a drug that is legally and medically identical to an expensive brand-name product without reinventing the wheel? The answer lies in a rigorous, highly regulated manufacturing process that balances science, law, and precision engineering.
This isn't just about copying a pill. It is about reverse-engineering complex chemistry while proving to regulators like the U.S. Food and Drug Administration (FDA) that the new version is safe, effective, and biologically equivalent. If you have ever wondered why your generic metformin costs a fraction of the price of the original but still lowers your blood sugar effectively, this breakdown explains the journey from raw chemical powder to the bottle on your pharmacy shelf.
The Legal Foundation: Why Generics Exist
To understand how generics are made, you first need to understand why they can be made at all. In 1984, Congress passed the Hatch-Waxman Act, officially known as the Drug Price Competition and Patent Term Restoration Act. Before this, if a company invented a new drug, they held a patent for years, preventing anyone else from making it. Once the patent expired, competitors could make copies, but they had to repeat every expensive clinical trial the original company did. That cost billions and took over a decade.
The Hatch-Waxman Act changed the game by creating the Abbreviated New Drug Application (ANDA) pathway. This allows generic manufacturers to skip the costly human safety trials because the brand-name drug has already proven it works. Instead, the generic maker only needs to prove two things: the drug contains the same active ingredient, and it behaves the same way in the human body. This cuts development time from 10-15 years to just 3-4 years and reduces costs from $2.6 billion to roughly $5-10 million per product. This legal shortcut is the engine that drives the entire generic manufacturing industry.
Step 1: Reverse Engineering the Reference Drug
The manufacturing process begins long before any machines start running. It starts with the Reference Listed Drug (RLD). This is the original brand-name medication. Generic manufacturers buy these pills and break them down in the lab. They analyze the molecular structure, the therapeutic effects, and the formulation characteristics.
The goal is to identify the Active Pharmaceutical Ingredient (API)-the actual medicine that treats your condition-and the excipients. Excipients are the inactive ingredients like binders, fillers, and coatings that hold the pill together or help it dissolve. While the API must be identical to the brand name, the excipients can differ. This is why your generic might be blue instead of white, or taste slightly different. However, these changes cannot affect how the drug works. Manufacturers use a framework called Quality by Design (QbD), established by the International Council for Harmonisation (ICH), to ensure that any change in materials does not compromise the drug's safety or efficacy.
Step 2: Formulation and Mixing
Once the formula is designed, the physical manufacturing begins. This happens in highly controlled environments. According to Current Good Manufacturing Practices (CGMPs), facilities must maintain specific temperatures (20-25°C) and humidity levels (45-65% RH). Cleanrooms are classified by ISO standards, ranging from Class 5 to Class 8, depending on how sensitive the stage of production is.
The process follows seven precise stages:
- Formulation: Designing the exact mix of API and excipients.
- Mixing and Granulation: Raw materials are combined into a consistent mixture. This is critical; if the mix isn't uniform, some pills will have too much medicine and others too little.
- Drying: Moisture is removed to ensure the final product is stable and won't degrade quickly.
- Compression and Encapsulation: Dry granules are pressed into tablets or filled into capsules. For tablets, weight variation is strictly monitored. The FDA requires that tablets under 130mg vary no more than ±5% in weight, while those between 130-324mg can vary up to ±7.5%.
- Coating: A protective layer is applied to mask taste, protect the drug from stomach acid, or control how fast it releases.
- Quality Control: Rigorous testing occurs at multiple stages.
- Packaging and Labeling: The final product is sealed and labeled with strict regulatory information.
A common headache in this phase, according to pharmaceutical engineers, is excipient variability. A slight change in the particle size of lactose from a supplier can ruin the tablet's hardness or dissolution profile. This is why manufacturers spend months validating their supply chains.

Step 3: Proving Bioequivalence
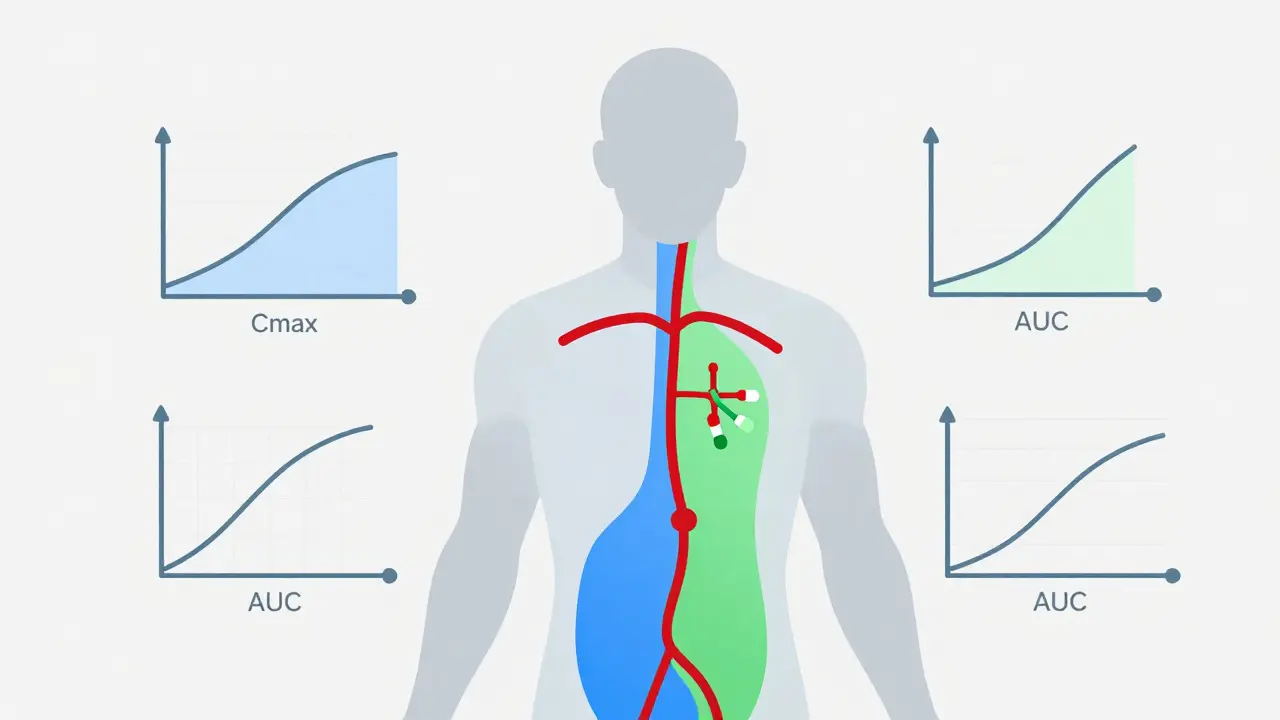
This is the most critical scientific hurdle. You cannot just make a pill and sell it. You must prove it is bioequivalent to the brand-name drug. Bioequivalence means the generic drug enters your bloodstream at the same rate and to the same extent as the original.
To prove this, manufacturers conduct pharmacokinetic studies involving 24-36 human subjects. These volunteers take both the brand-name drug and the generic drug (usually in a crossover design where they take one, wash out, then take the other). Blood samples are taken to measure two key metrics:
- Cmax: The maximum concentration of the drug in the blood.
- AUC: The area under the curve, representing total exposure to the drug over time.
FDA guidelines require that the generic's Cmax and AUC fall within an 80%-125% confidence interval of the brand-name drug's parameters. If the generic is absorbed too slowly or too quickly, it fails. This statistical range ensures that for the vast majority of patients, there is no clinically meaningful difference in effect. For narrow therapeutic index drugs-where small dosage changes matter greatly-the scrutiny is even tighter.
Step 4: Regulatory Approval and Inspection
With the data collected, the manufacturer submits the ANDA application. This document is massive, often containing 5,000 to 10,000 pages of technical data, including hundreds of analytical methods and batch records. The FDA reviews this submission, which typically takes about 17 months for standard applications, though GDUFA IV goals aim to review 90% of original ANDAs within 10 months.
But paperwork isn't enough. The FDA inspects the manufacturing facility. Inspectors look for compliance with CGMPs. Common deficiencies cited in warning letters include inadequate investigation of out-of-specification results (found in 37% of warnings) and insufficient process validation (29%). If a facility passes inspection and the ANDA is approved, the generic can hit the market. The labeling must match the brand-name drug exactly, except for the manufacturer's name and certain trademarked terms.
Complex Generics: The Harder Challenge
Not all generics are created equal. Simple tablets are straightforward. But "complex generics"-like inhalers, topical creams, nasal sprays, and modified-release formulations-pose significant challenges. For these products, traditional bioequivalence testing may not be enough. For example, matching the skin permeation profile of a topical corticosteroid can take years. One case study noted a manufacturer spent seven years and $47 million developing a generic version of a topical steroid because matching the reference product's performance was so difficult.
The FDA has launched initiatives to address this, publishing over 127 product-specific guidances for complex generics by late 2023. These products face fewer competitors (often 2-5 versus 15-20 for simple generics) and maintain higher prices longer, making them a strategic focus for many manufacturers.
| Attribute | Brand-Name Drug | Generic Drug |
|---|---|---|
| Development Time | 10-15 years | 3-4 years |
| Estimated Cost | $2.6 billion | $5-10 million |
| Clinical Trials | Extensive Phase I-III trials required | Bioequivalence studies only |
| Regulatory Pathway | New Drug Application (NDA) | Abbreviated New Drug Application (ANDA) |
| Price Impact | High (patent protection) | Low (80-85% less than brand) |
Quality Control and Future Trends
Manufacturing doesn't stop after approval. Post-approval monitoring ensures consistency. Every batch must meet exact specifications for identity, strength, purity, and quality. Stability testing requires at least 12 months of real-time data before initial approval, and ongoing tests monitor degradation over time.
The industry is also evolving. Continuous manufacturing, where drugs are produced in a steady flow rather than discrete batches, is gaining traction. The FDA has approved 17 continuous manufacturing facilities for generics as of 2023. This technology reduces production time from weeks to hours and improves quality consistency, with some companies achieving 99.98% batch acceptance rates compared to 95% with traditional methods. Additionally, AI-driven quality control systems are being piloted to reduce visual inspection errors, marking a shift toward smarter, more efficient production lines.
Despite occasional recalls due to CGMP violations, the system works. Surveys show that 89% of pharmacists report high confidence in generic drug quality. The generic model remains one of the most successful public health initiatives in modern history, ensuring that life-saving medications remain accessible to millions who otherwise could not afford them.
Are generic drugs really as effective as brand-name drugs?
Yes. By law, generic drugs must contain the same active ingredient, strength, dosage form, and route of administration as the brand-name drug. They must also demonstrate bioequivalence, meaning they perform identically in the body within a strict statistical margin (80%-125%). The FDA confirms that generics provide the same clinical benefit and safety profile as their brand-name counterparts.
Why do generic drugs look different from brand-name drugs?
Generic drugs may differ in color, shape, flavor, and inactive ingredients (excipients). This is allowed because these factors do not affect the drug's therapeutic action. Trademark laws also prevent generics from looking exactly like the brand-name product to avoid consumer confusion. As long as the active ingredient and bioavailability are the same, the appearance differences are purely cosmetic.
What is the ANDA process?
ANDA stands for Abbreviated New Drug Application. It is the regulatory pathway used to approve generic drugs. Unlike new brand-name drugs, which require extensive clinical trials, ANDAs rely on the existing safety and efficacy data of the brand-name drug. The generic manufacturer only needs to prove bioequivalence and demonstrate that their manufacturing processes meet strict quality standards.
How long does it take to manufacture a generic drug?
The entire development and approval process for a generic drug typically takes 3 to 4 years. This includes formulation development, bioequivalence studies, ANDA submission, and FDA review. The actual physical manufacturing of a batch takes days to weeks, depending on complexity, but the pre-market timeline is dominated by regulatory and testing phases.
What are complex generics?
Complex generics are drugs that are difficult to replicate due to their delivery system or formulation, such as inhalers, topical creams, patches, or modified-release tablets. Demonstrating bioequivalence for these products is harder than for simple oral tablets, often requiring specialized testing methods and longer development times. They represent a growing segment of the generic market with fewer competitors.

