Imagine spending months and millions of dollars on a clinical trial, only to have the bioequivalence standards you followed result in a total failure because the drug was just too "noisy." For drugs with high variability, the traditional 2x2 crossover design-the gold standard for decades-often hits a wall. When the drug's levels swing wildly between different people, a standard study would require hundreds of participants to reach statistical significance, which is practically impossible for most sponsors. This is where replicate study designs step in. They aren't just an alternative; for certain medications, they are the only viable path to regulatory approval.
What exactly are replicate study designs?
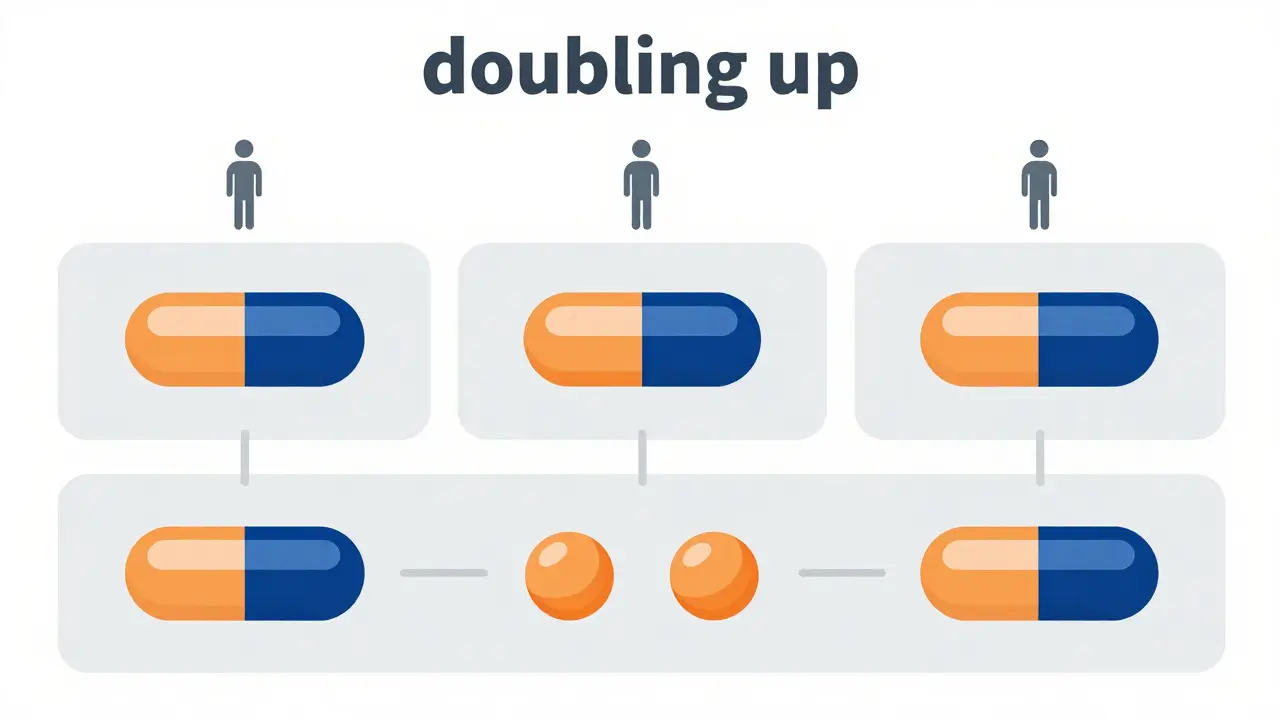
A replicate study design is a specialized methodology where subjects receive multiple doses of the test product, the reference product, or both, across several treatment periods. Unlike a standard design where you give the drug once and move on, replicate designs "double up" on dosing. This allows researchers to isolate and measure the within-subject variability-the natural fluctuation of a drug's concentration in the same person over time-separately from the variability between different people.
This distinction is critical for highly variable drugs (HVDs). A drug is generally classified as highly variable when its intra-subject coefficient of variation (ISCV) exceeds 30%. When you're dealing with that level of noise, the U.S. Food and Drug Administration (FDA) and the European Medicines Agency (EMA) allow for Reference-Scaled Average Bioequivalence (RSABE). Essentially, if the reference drug is naturally wild, the regulators "scale" the acceptance limits to be a bit wider, provided you can prove exactly how variable that reference drug is using a replicate design.
Breaking down the three main design types
Not all replicate designs are created equal. Depending on your drug's profile and the regulatory body you're answering to, you'll likely choose one of these three paths:
- Full Replicate Designs: These are the most robust. In a four-period sequence (like TRRT or RTRT), subjects get both the test and reference products twice. In three-period versions (TRT or RTR), they get one twice and the other once. Because you have repeats for both, you can calculate the variability for both the test and reference formulations. For Narrow Therapeutic Index (NTI) drugs, the FDA usually mandates the four-period version to ensure maximum precision.
- Partial Replicate Designs: These use three-period sequences (such as TRR or RTR). The key difference here is that the test product is only administered once. This design allows you to estimate the variability of the reference product only, which is sufficient for RSABE analysis. It's faster and cheaper, but offers less data on the test product's behavior.
- Standard 2x2 Crossover: While not a replicate design, it's the baseline. Subjects get T then R, or R then T. While simple, it cannot separate within-subject variability, making it a recipe for failure when the ISCV is over 30%.
| Feature | Standard 2x2 | Partial Replicate | Full Replicate (4-period) |
|---|---|---|---|
| Dosing Periods | 2 | 3 | 4 |
| Reference Variability Est. | No | Yes | Yes |
| Test Variability Est. | No | No | Yes |
| Typical Sample Size (HVD) | 100+ subjects | 24-48 subjects | 24-72 subjects |
| Regulatory Risk (HVD) | Very High | Moderate | Low |
The math of sample size: Why it matters
The real-world value of these designs is found in the recruitment numbers. Let's look at a concrete example: imagine a drug with an ISCV of 50% and a formulation difference (FD) of 10%. In a standard crossover study, you would need roughly 108 subjects to achieve statistical power. With a replicate design, that number drops to just 28 subjects. That is a massive reduction in cost, time, and the number of human volunteers needed.
However, this efficiency comes with a trade-off. You are asking each subject to come back for more visits. This increases the "subject burden," which naturally leads to higher dropout rates. Industry data suggests an average dropout rate of 15-25% for multi-period studies. If you need 24 evaluable subjects, don't recruit 24; recruit 30 or 32 to account for the people who will inevitably decide they've had enough of the clinic.

Avoiding the common pitfalls
Even with a great design, things can go wrong. A common mistake is neglecting the washout period. Because replicate designs involve more doses, the time between treatments must be strictly managed to ensure the drug from the first period is completely gone before the second begins. If the drug has a long half-life, this can stretch a study's duration by weeks or months.
Then there is the statistical hurdle. You can't just plug this data into a basic spreadsheet. You need specialized software like Phoenix WinNonlin or the replicateBE package in R. Many analysts spend 80 to 120 hours just training on how to handle these mixed-effects models correctly. If you pick the wrong statistical model, the FDA or EMA will likely reject the submission, regardless of how good the actual drug performance was.
Choosing the right design for your drug
How do you decide which path to take? It usually comes down to a simple rule of thumb based on expected variability:
- ISCV < 30%: Stick with the standard 2x2 crossover. It's the fastest and most accepted route.
- ISCV between 30% and 50%: The three-period full replicate (TRT/RTR) is often the "sweet spot," balancing statistical power with operational feasibility.
- ISCV > 50% or NTI Drugs: Go for the four-period full replicate (TRRT/RTRT). When the stakes are high-like with Warfarin Sodium-the regulators want a complete picture of both test and reference variability.
We're also seeing a shift toward adaptive designs. Some sponsors start with a replicate study but include a plan to switch to standard analysis if the early data shows the variability is lower than expected. This reduces the risk of over-engineering the study while still providing a safety net.
Why are replicate designs required for highly variable drugs?
Standard 2x2 designs cannot separate the variability within a single subject from the variability between different subjects. For drugs with an ISCV over 30%, this makes it nearly impossible to prove bioequivalence without an impractically large sample size. Replicate designs allow for "reference scaling," where the acceptance limits are adjusted based on the reference drug's own variability.
What is the difference between a partial and a full replicate design?
A full replicate design provides multiple doses of both the test and reference products, allowing the calculation of variability for both. A partial replicate design provides multiple doses of the reference product but only one dose of the test product, meaning only the reference product's variability can be estimated.
Which software is best for analyzing replicate BE studies?
The industry standard is Phoenix WinNonlin for general pharmacokinetic analysis. For those using open-source tools, the R package 'replicateBE' is widely used and recognized for its ability to handle RSABE calculations according to regulatory guidelines.
Do the FDA and EMA have the same requirements for these studies?
While both agencies accept RSABE, there are slight differences. For example, the FDA has historically been more prescriptive about four-period designs for NTI drugs, while the EMA has shown slightly more flexibility with three-period designs. Harmonization is ongoing through the ICH, but it's always best to check the specific Product-Specific Guidances (PSGs) for each agency.
How do I handle high dropout rates in multi-period studies?
Because subjects must return for 3 or 4 periods, dropout is common (often 15-25%). The best approach is over-recruitment-recruiting 20-30% more subjects than the statistical power calculation requires-and ensuring a strong subject retention program to keep volunteers engaged.
The distinction between within-subject and between-subject variability is indeed the cornerstone of these designs. It is also worth noting that for certain drugs, the choice of the washout period can be influenced by the specific metabolic pathway of the drug, which further complicates the operational logistics of a four-period study.
Everyone knows that 2x2 is basically useless for high noise drugs. It's just common sense.
american standards are the only ones that actually matter in this game and if you aren't following fda a you are just playing around with toys
statistcs are just a way to hide the truth with numbers anyway... who knows what the pharma guys are actually hiding in those mixed-effects modls
Scaling limits is just a loophole. They're hiding the danger.
Exactly. The whole 'highly variable' excuse is just a way for big pharma to push through generics that aren't actually equivalent. They manipulate the RSABE numbers to make things fit.
I just like that it helps people get their meds faster! 😊✨
listen up juniors if you dont use r package replicateBE you are wasting time stop using spreadsheets and get real
WinNonlin is too expensive. R is better
Oh sure, let's just tell the volunteers to come back for a fourth time. I'm sure they'll be absolutely thrilled to spend another week in a sterile clinic eating bland food for the sake of a p-value. Truly a dream scenario for everyone involved.
This is basically Bio-Stats 101. If you don't get why a partial replicate is cheaper, you shouldn't be in the industry. It's simple logic. You save money because you don't waste test drug on the second dose. Period.
Keep pushing through those study designs! It's tough work but it really makes a difference for patients who need these meds. You've got this!
Too long. TL;DR is basically just use more doses if the drug is noisy.
It's so important to remember the human side of this. Those volunteers are doing a great thing for science. I hope the clinics are taking good care of them during those long multi-period stays. Maybe a bit of comfort would help the dropout rates!
I can imagine how stressful it is for the analysts when the data comes back and the ISCV is slightly over 30% and they have to pivot the whole design. It's a lot of pressure to get those regulatory submissions right the first time.
The part about over-recruiting is a lifesaver because people really do just disappear from these trials for the weirdest reasons. I've seen folks drop out because they didn't like the pillows in the clinic. It's a strange business.
The shift toward adaptive designs is probably the smartest move here. It gives a safety net for the sponsor while still being rigorous.
Using Phoenix WinNonlin is definitely the safest bet for FDA approval even if the learning curve is a nightmare.
The mention of Warfarin is a great example because NTI drugs are just in a different league of risk.
One must also consider the cost of the drug itself when choosing between partial and full replicates.
If the test drug is incredibly expensive, a partial design isn't just faster, it's a financial necessity.
The washout period is where most of the operational headaches actually happen.
It's funny how a few days of extra washout can throw off a whole clinic's schedule for a month.
The math on the sample size reduction from 108 to 28 is just staggering when you think about the cost per subject.
It really highlights why these advanced methods are a game changer.
I appreciate the breakdown of the three design types.
It makes the complex regulatory environment much easier to navigate.
Most people would just try the 2x2 and fail miserably.
Good luck to everyone currently battling with their BE data!